Que TDP-43 está implicado en el 95% de los casos de ELA y en la mitad de los de demencia frontotemporal (DTP) se sabe en cuanto se empieza a leer sobre el tema. Sin embargo, sigue sin estar claro cómo está implicado. De forma habitual tiene una función dentro del núcleo uniéndose a ácidos nucleicos (ADN y ARN). En muestras de tejido afectado por la ELA, TDP-43 se encuentra fuera del núcleo, formando cúmulos. En trabajos de los últimos meses se descubrió que estos cúmulos, por sí mismos, estaban implicados en causar neurodegeneración (Altman et al., 2021). Sin embargo, dos trabajos simultáneos en el University College de Londres y la Escuela de Medicina de Stanford liderados por el Dr. Pietro Fratta y el Dr. Aaron Gitler respectivamente han ido más allá. Evidentemente, si TDP-43 se encuentra en agregados fuera del núcleo, ya no estará ejerciendo su función normal pero, ¿cómo de relevante puede ser su papel?
Para poder generar las proteínas que llevan a cabo las funciones en las células existe un proceso con varios pasos. Las “instrucciones” para generar proteínas se guardan en el ADN formado por un conjunto de sus subunidades, los nucleótidos, de los que hay solo 4 tipos diferentes que se combinan. Sin embargo, no todo el ADN va a dar lugar a proteínas. Es necesario hacer un negativo de la secuencia de ADN, que necesitamos, lo que llamamos ARN mensajero (porque lleva el mensaje sobre cuál es el ADN que va a dar una proteína) (1). El código genético es una clave que nos va a permitir el proceso de traducción del ARN. Cada tres subunidades que forman el ARN, se van a traducir en un aminoácido, es decir, las piezas que forman las proteínas. Pero no se usa directamente el ARNm ya que primero necesita un procesamiento que llamamos splicing (2). Hay partes del ARNm que van a ser seleccionadas (exones) y otras no se incluyen en la forma final del ARNm (intrones). Pero además, no todas las partes que pueden ser seleccionadas, se seleccionan siempre. El splicing será el proceso de selección para incluir todos los exones requeridos porque se reconocerán los sitios del ARNm habituales para hacer el splicing. Este estudio explica cómo TDP-43 es importante durante el splicing de UNC13A porque impide el reconocimiento de sitios de splicing no habituales (crípticos) (3). En una situación sana, el splicing será normal (4), dando una proteína UNC13A normal (5). Su función es muy relevante en la liberación del neurotransmisor en las sinapsis.
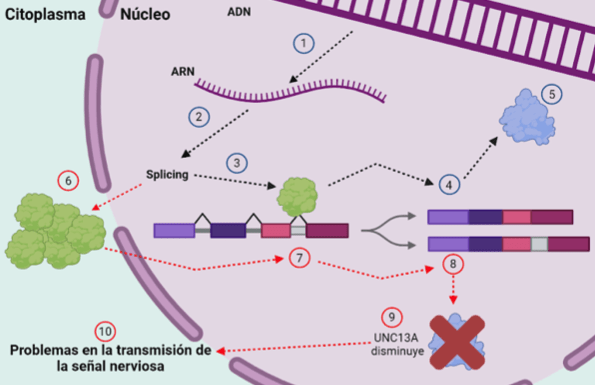
En la ELA, un 95% de los afectados aproximadamente, sufren alteraciones en TDP-43, que sale del núcleo y forma cúmulos (6). En artículos anteriores publicamos cómo se había descubierto que por sí mismos inducen neurodegeneración. Pero además, una importante escasez de TDP-43 en el núcleo le impide realizar su función habitual, lo que es propio de la fase final de la enfermedad (7). Esto provoca que un exón de más se incluya en el ARN final procesado (8). Este exón extra tiene una señal que provoca la detención prematura de la traducción, dando lugar a una proteína UNC13A incompleta y no funcional (9) con los consecuentes problemas en la liberación del neurotransmisor y en la transmisión de la señal nerviosa que conlleva su falta (10).
Existe un tipo de variación en el ADN llamada SNP, en la que cambia un único nucleótido de la secuencia. Algunos SNP de UNC13A han sido asociados a una ELA que avanza más rápidamente. En estos trabajos demostraron que estos SNP estaban relacionados con el proceso de splicing que hemos explicado anteriormente. Estos dos trabajos en conjunto han demostrado que los SNP asociados a ELA están relacionados con el exón críptico. Solo tener estas variantes o alelos de riesgo no son suficiente para causar la inclusión del exón. Pero sí que van a exacerbar la inclusión del exón extra, incluso con una pérdida parcial de TDP-43. Esto explicaría que las personas con estas variantes tengan una progresión más rápida. Son resultados que señalan al exón extra como posible diana terapéutica de una terapia capaz de evitar el splicing erróneo y que sería aplicable al 97% de los pacientes de ELA y a la mitad de los casos de DFT.
Referencia:
Brown, A. L., Wilkins, O. G., Keuss, M. J., Hill, S. E., Zanovello, M., Lee, W. C., Bampton, A., Lee, F., Masino, L., Qi, Y. A., Bryce-Smith, S., Gatt, A., Hallegger, M., Fagegaltier, D., Phatnani, H., NYGC ALS Consortium, Newcombe, J., Gustavsson, E. K., Seddighi, S., Reyes, J. F., … Fratta, P. (2022). TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature, 603(7899), 131–137. https://doi.org/10.1038/s41586-022-04436-3
Ma, X. R., Prudencio, M., Koike, Y., Vatsavayai, S. C., Kim, G., Harbinski, F., Briner, A., Rodriguez, C. M., Guo, C., Akiyama, T., Schmidt, H. B., Cummings, B. B., Wyatt, D. W., Kurylo, K., Miller, G., Mekhoubad, S., Sallee, N., Mekonnen, G., Ganser, L., Rubien, J. D., … Gitler, A. D. (2022). TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature, 603(7899), 124–130. https://doi.org/10.1038/s41586-022-04424-7



